
Electrophilic aromatic substitution
Electrophilic aromatic substitution (SEAr) is an organic reaction in which an atom that is attached to an aromatic system (usually hydrogen) is replaced by an electrophile. Some of the most important electrophilic aromatic substitutions are aromatic nitration, aromatic halogenation, aromatic sulfonation, alkylation Friedel–Crafts reaction and acylation Friedel–Crafts reaction.[1]
Illustrative reactions
[edit]The most widely practised example of this reaction is the ethylation of benzene.

Approximately 24,700,000 tons were produced in 1999.[2] (After dehydrogenation and polymerization, the commodity plastic polystyrene is produced.) In this process, acids are used as catalyst to generate the incipient carbocation. Many other electrophilic reactions of benzene are conducted, although on a much smaller scale; they are valuable routes to key intermediates. The nitration of benzene is achieved via the action of the nitronium ion as the electrophile. The sulfonation with fuming sulfuric acid gives benzenesulfonic acid. Aromatic halogenation with bromine, chlorine, or iodine gives the corresponding aryl halides. This reaction is typically catalyzed by the corresponding iron or aluminum trihalide.

The Friedel–Crafts reaction can be performed either as an acylation or as an alkylation. Often, aluminium trichloride is used, but almost any strong Lewis acid can be applied. For the acylation reaction a stoichiometric amount of aluminum trichloride is required.


Reaction mechanism
[edit]The overall reaction mechanism, denoted by the Hughes–Ingold mechanistic symbol SEAr,[3] begins with the aromatic ring attacking the electrophile E+ (2a). This step leads to the formation of a positively charged and delocalized cyclohexadienyl cation, also known as an arenium ion, Wheland intermediate, or arene σ-complex (2b). Many examples of this carbocation have been characterized, but under normal operating conditions these highly acidic species will donate the proton attached to the sp3 carbon to the solvent (or any other weak base) to reestablish aromaticity. The net result is the replacement of H by E in the aryl ring (3).
Occasionally, other electrofuges (groups that can leave without their electron pair) beside H+ will depart to reestablish aromaticity; these species include silyl groups (as SiR3+), the carboxy group (as CO2 + H+), the iodo group (as I+), and tertiary alkyl groups like t-butyl (as R+). The capacity of these types of substituents to leave is sometimes exploited synthetically, particularly the case of replacement of silyl by another functional group (ipso attack). However, the loss of groups like iodo or alkyl is more often an undesired side reaction.

Effect of substituent groups
[edit]
Both the regioselectivity—the diverse arene substitution patterns—and the speed of an electrophilic aromatic substitution are affected by the substituents already attached to the benzene ring. In terms of regioselectivity, some groups promote substitution at the ortho or para positions, whereas other groups favor substitution at the meta position. These groups are called either ortho–para directing or meta directing, respectively. In addition, some groups will increase the rate of reaction (activating) while others will decrease the rate (deactivating). While the patterns of regioselectivity can be explained with resonance structures, the influence on kinetics can be explained by both resonance structures and the inductive effect.
Reaction rate
[edit]Substituents can generally be divided into two classes regarding electrophilic substitution: activating and deactivating towards the aromatic ring. Activating substituents or activating groups stabilize the cationic intermediate formed during the substitution by donating electrons into the ring system, by either inductive effect or resonance effects. Examples of activated aromatic rings are toluene, aniline and phenol.
The extra electron density delivered into the ring by the substituent is not distributed evenly over the entire ring but is concentrated on atoms 2, 4 and 6, so activating substituents are also ortho/para directors (see below).
On the other hand, deactivating substituents destabilize the intermediate cation and thus decrease the reaction rate by either inductive or resonance effects. They do so by withdrawing electron density from the aromatic ring. The deactivation of the aromatic system means that generally harsher conditions are required to drive the reaction to completion. An example of this is the nitration of toluene during the production of trinitrotoluene (TNT). While the first nitration, on the activated toluene ring, can be done at room temperature and with dilute acid, the second one, on the deactivated nitrotoluene ring, already needs prolonged heating and more concentrated acid, and the third one, on very strongly deactivated dinitrotoluene, has to be done in boiling concentrated sulfuric acid. Groups that are electron-withdrawing by resonance decrease the electron density especially at positions 2, 4 and 6, leaving positions 3 and 5 as the ones with comparably higher reactivity, so these types of groups are meta directors (see below). Halogens are electronegative, so they are deactivating by induction, but they have lone pairs, so they are resonance donors and therefore ortho/para directors.
Ortho/para directors
[edit]Groups with unshared pairs of electrons, such as the amino group of aniline, are strongly activating (some time deactivating also in case of halides) and ortho/para-directing by resonance. Such activating groups donate those unshared electrons to the pi system, creating a negative charge on the ortho and para positions. These positions are thus the most reactive towards an electron-poor electrophile. This increased reactivity might be offset by steric hindrance between activating group and electrophile but on the other hand there are two ortho positions for reaction but only one para position. Hence the final outcome of the electrophilic aromatic substitution is difficult to predict, and it is usually only established by doing the reaction and observing the ratio of ortho versus para substitution.

resonance structures for ortho attack of an electrophile on aniline

In addition to the increased nucleophilic nature of the original ring, when the electrophile attacks the ortho and para positions of aniline, the nitrogen atom can donate electron density to the pi system (forming an iminium ion), giving four resonance structures (as opposed to three in the basic reaction). This substantially enhances the stability of the cationic intermediate.

Resonance structures for para attack

When the electrophile attacks the meta position, the nitrogen atom cannot donate electron density to the pi system, giving only three resonance contributors. This reasoning is consistent with low yields of meta-substituted product.

resonance structures for meta attack of an electrophile on aniline

Other substituents, such as the alkyl and aryl substituents, may also donate electron density to the pi system; however, since they lack an available unshared pair of electrons, their ability to do this is rather limited. Thus, they only weakly activate the ring and do not strongly disfavor the meta position.
Directed ortho metalation is a special type of EAS with special ortho directors.
Meta directors
[edit]Non-halogen groups with atoms that are more electronegative than carbon, such as a carboxylic acid group (-CO2H), withdraw substantial electron density from the pi system. These groups are strongly deactivating groups. Additionally, since the substituted carbon is already electron-poor, any structure having a resonance contributor in which there is a positive charge on the carbon bearing the electron-withdrawing group (i.e., ortho or para attack) is less stable than the others. Therefore, these electron-withdrawing groups are meta directing because this is the position that does not have as much destabilization.
The reaction is also much slower (a relative reaction rate of 6×10−8 compared to benzene) because the ring is less nucleophilic.
Steric effects
[edit]Although discussions of directing groups usually focus on electronic effects (e.g. EWG vs EDGs), steric effect can prove influential. Thus, nitration of toluene gives approximately 2:1 ortho vs para-nitrotoluene. In the case of tert-butylbenzene, however, the selectivity is reversed:73% of the product is 4-nitro-tert-butybenzene]].[4]
Reaction on pyridine
[edit]Compared to benzene, the rate of electrophilic substitution on pyridine is much slower, due to the higher electronegativity of the nitrogen atom. Additionally, the nitrogen in pyridine easily gets a positive charge either by protonation (from nitration or sulfonation) or Lewis acids (such as AlCl3) used to catalyze the reaction. This makes the reaction even slower by having adjacent formal charges on carbon and nitrogen or 2 formal charges on a localised atom. Doing an electrophilic substitution directly in pyridine is nearly impossible.
In order to do the reaction, they can be made by 2 possible reactions, which are both indirect.
One possible way to do a substitution on pyridine is nucleophilic aromatic substitution. Even with no catalysts, the nitrogen atom, being electronegative, can hold the negative charge by itself. Another way is to do an oxidation before the electrophilic substitution. This makes pyridine N-oxide, which due to the negative oxygen atom, makes the reaction faster than pyridine, and even benzene. The oxide then can be reduced to the substituted pyridine.
Ipso attack
[edit]The attachment of an entering group to a position in an aromatic compound already carrying a substituent group (other than hydrogen). The entering group may displace that substituent group but may also itself be expelled or migrate to another position in a subsequent step. The term 'ipso-substitution' is not used, since it is synonymous with substitution.[5] A classic example is the reaction of salicylic acid with a mixture of nitric and sulfuric acid to form picric acid. The nitration of the 2 position involves the loss of CO2 as the leaving group. Desulfonation in which a sulfonyl group is substituted by a proton is a common example. See also Hayashi rearrangement. In aromatics substituted by silicon, the silicon reacts by ipso substitution.
Five membered heterocycles
[edit]Compared to benzene, furans, thiophenes, and pyrroles are more susceptible to electrophilic attack. These compounds all contain an atom with an unshared pair of electrons (oxygen, sulfur, or nitrogen) as a member of the aromatic ring, which substantially stabilizes the cationic intermediate. Examples of electrophilic substitutions to pyrrole are the Pictet–Spengler reaction and the Bischler–Napieralski reaction.
Asymmetric electrophilic aromatic substitution
[edit]Electrophilic aromatic substitutions with prochiral carbon electrophiles have been adapted for asymmetric synthesis by switching to chiral Lewis acid catalysts especially in Friedel–Crafts type reactions. An early example concerns the addition of chloral to phenols catalyzed by aluminium chloride modified with (–)-menthol.[6] A glyoxylate compound has been added to N,N-dimethylaniline with a chiral bisoxazoline ligand–copper(II) triflate catalyst system also in a Friedel–Crafts hydroxyalkylation:[7]

Asymmetric Friedel–Crafts hydroxyalkylation

In another alkylation N-methylpyrrole reacts with crotonaldehyde catalyzed by trifluoroacetic acid modified with a chiral imidazolidinone:[8]
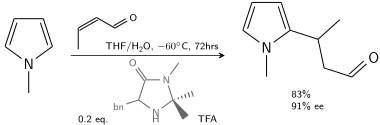
Friedel Crafts Asymmetric Addition To Pyrrole

Indole reacts with an enamide catalyzed by a chiral BINOL derived phosphoric acid:[9]
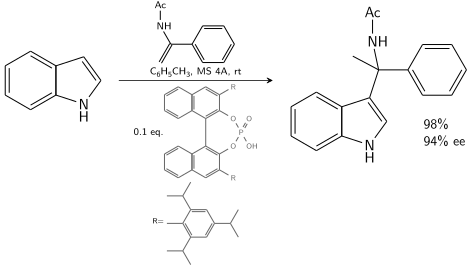
Friedel Crafts Alkylation Indole Asymmetric

In the presence of 10–20 % chiral catalyst, 80–90% ee is achievable.
Other reactions
[edit]- Other reactions that follow an electrophilic aromatic substitution pattern are a group of aromatic formylation reactions including the Vilsmeier–Haack reaction, the Gattermann Koch reaction and the Reimer–Tiemann reaction.
- Other electrophiles are aromatic diazonium salts in diazonium couplings, carbon dioxide in the Kolbe–Schmitt reaction and activated carbonyl groups in the Pechmann condensation, hydroxycarbenium ion in the Blanc chloromethylation via an intermediate (hydroxymethyl)arene (benzyl alcohol), chloryl cation (ClO3+) for electrophilic perchlorylation.
- In the multistep Lehmstedt–Tanasescu reaction, one of the electrophiles is a N-nitroso intermediate.
- In the Tscherniac–Einhorn reaction (named after Joseph Tscherniac and Alfred Einhorn) the electrophile is a N-methanol derivative of an amide[10][11]
See also
[edit]References
[edit]- ^ Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 978-0-471-72091-1
- ^ Vincent A. Welch, Kevin J. Fallon, Heinz-Peter Gelbke "Ethylbenzene" Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005. doi:10.1002/14356007.a10_035.pub2
- ^ Gawley, Robert E. (1999-06-04). "A proposal for (slight) modification of the Hughes–Ingold mechanistic descriptors for substitution reactions". Tetrahedron Letters. 40 (23): 4297–4300. doi:10.1016/S0040-4039(99)00780-7. ISSN 0040-4039.
- ^ Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, p. 671, ISBN 978-0-471-72091-1
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "ipso-attack". doi:10.1351/goldbook.I03251
- ^ Asymmetric electrophilic substitution on phenols in a Friedel–Crafts hydroxyalkylation. Enantioselective ortho-hydroxyalkylation mediated by chiral alkoxyaluminum chlorides Franca Bigi, Giovanni Casiraghi, Giuseppe Casnati, Giovanni Sartori, Giovanna Gasparri Fava, and Marisa Ferrari Belicchi J. Org. Chem.; 1985; 50(25) pp 5018–5022; doi:10.1021/jo00225a003
- ^ Catalytic Enantioselective Friedel–Crafts Reactions of Aromatic Compounds with Glyoxylate: A Simple Procedure for the Synthesis of Optically Active Aromatic Mandelic Acid Esters Nicholas Gathergood, Wei Zhuang, and Karl Anker Jrgensen J. Am. Chem. Soc.; 2000; 122(50) pp 12517–12522; (Article) doi:10.1021/ja002593j
- ^ New Strategies in Organic Catalysis: The First Enantioselective Organocatalytic Friedel–Crafts Alkylation Nick A. Paras and David W. C. MacMillan J. Am. Chem. Soc.; 2001; 123(18) pp. 4370–4371; (Communication) doi:10.1021/ja015717g
- ^ Chiral Brønsted Acid Catalyzed Enantioselective Friedel–Crafts Reaction of Indoles and a-Aryl Enamides: Construction of Quaternary Carbon Atoms Yi-Xia Jia, Jun Zhong, Shou-Fei Zhu, Can-Ming Zhang, and Qi-Lin Zhou Angew. Chem. Int. Ed. 2007, 46, 5565 –5567 doi:10.1002/anie.200701067
- ^ Verfahren zur Darstellung von Benzylphtalimiden Joseph Tscherniac German Patent 1902, DE-134,979
- ^ Ueber die N-Methylolverbindungen der Säureamide [Erste Abhandlung.] Alfred Einhorn, Eduard Bischkopff, Bruno Szelinski, Gustav Schupp, Eduard Spröngerts, Carl Ladisch, and Theodor Mauermayer Liebigs Annalen 1905, 343, pp. 207–305 doi:10.1002/jlac.19053430207